
General introduction to reaction-diffusion equations and GPGMP
GPGMP provides an implementation of a spatially-resolved stochastic simulation algorithm on general-purpose graphics processing units (GPGPUs). This algorithm, designed to solve reaction-diffusion equations on a mesoscopic level, has widespread applications in computational biology, molecular chemistry and social systems research. For our implementation, we use the Gillespie multi-particle method, which is well-suited for GPGPUs. In preliminary tests, we could obtain speed ups of around 1-2 orders of magnitudes as compared to standard implementations on CPUs.
In many applications, most notably in a biological and chemical context, the number of participating agents is small and the inherently stochastic nature of the problem requires a microscopic description of the system, in which the position of every species is known at all times. The macroscopic diffusion equation can be regained by appropriate statistic averaging.
A microscopic approach, however, is often cumbersome and computer simulations to obtain realizations of reaction-diffusion networks are quite costly. Furthermore, it is often not necessary to distinguish between individual entities and a mesoscopic approach is sufficient. In a mesoscopic description, the domain of interest is divided into small subvolumes of a convenient granularity, which are assumed to be perfectly mixed. Instead of tracing the position of each individual particle through time, one then collectively keeps track of the number of particles in each subvolume.
All algorithms in the previous paragraph compute exact realizations of stochastic reaction-diffusion systems. The disadvantage of these methods is their high computational cost, in particular, if a fine granularity is required and the number of subvolumes is high. Furthermore, they do not lend themselves well for data-parallel implementations. However, if one is willing to sacrifice accuracy for speed, deterministic-stochastic hybrid algorithms provide an efficient alternative. In these methods, the diffusion part is separated from the reaction part and treated deterministically. The Gillespie multiparticle method (GMP) (Rodriguez et al., 2006) is one example of this class of algorithms.
GMP handles the diffusion part of the reaction-diffusion network trough a multiparticle lattice gas automaton (Chopard et al., 1994) and the reaction part via Gillespie's direct method (Gillespie, 1977). The time step is chosen globally according to the diffusion constants of each species and the dimensions of each lattice cell. In each iteration, all reactions are performed independently in each subvolume. At the end, all particles are diffused according to the cellular automaton rule.
We present a data-parallel implementation of GMP on GPGPUs (GPGMP). Since all cells are treated independently during each time step, our approach is straight-forward. Each thread works independently on exactly one subvolume. Global synchronization is only required at the end of the diffusion time step to exchange information about particles moving in between neighbouring cells. We refer the reader to Vigelius et al. (2010) for implementation details.
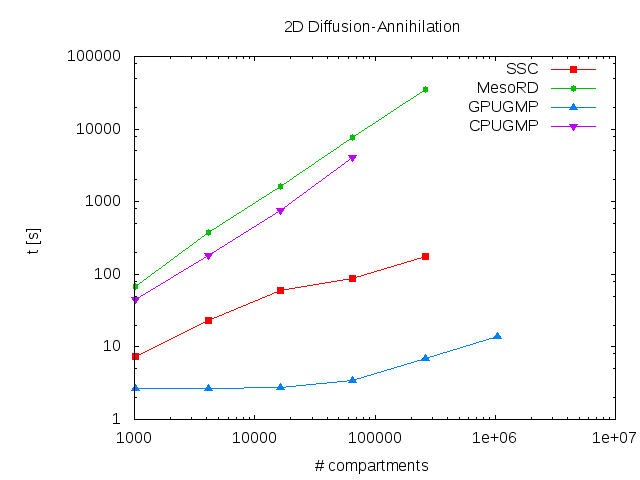
GPGMP allows researchers to efficiently simulate spatially-inhomogeneous reaction-diffusion networks. Even if no access to designated GPGPU clusters is available, GPGMP can still provide considerable speed gains on built-in GPGPU cards on workstations (see the figure above for a comparison of several algorithms). Furthermore, we provide free access to the Monash Sun Grid GPGMP cluster through our easy-to-use web interface to GPGMP, Inchman.
If you find this software useful, we kindly ask you to reference Vigelius et al. (2010) in your publication.
For more information, please see the GPGMP Developer Site.
For more information, please see the Inchman Tutorial.