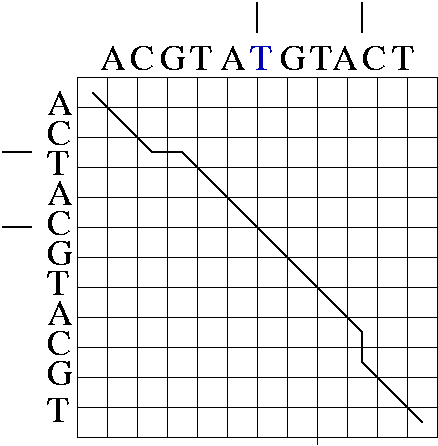
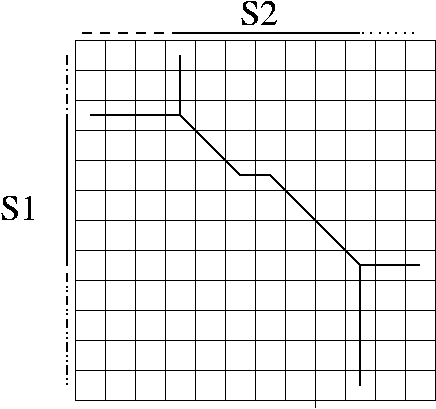
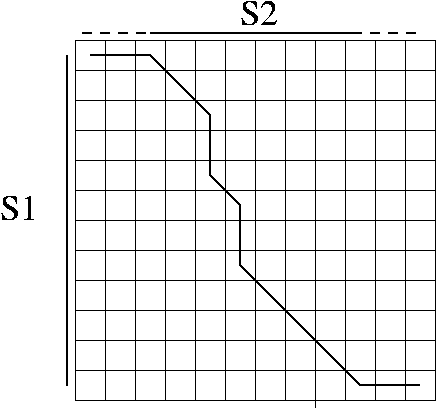
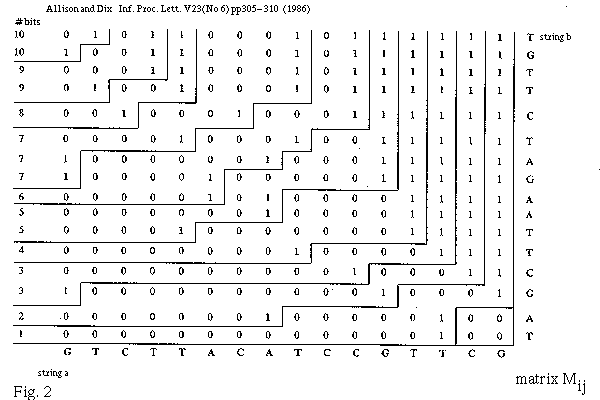
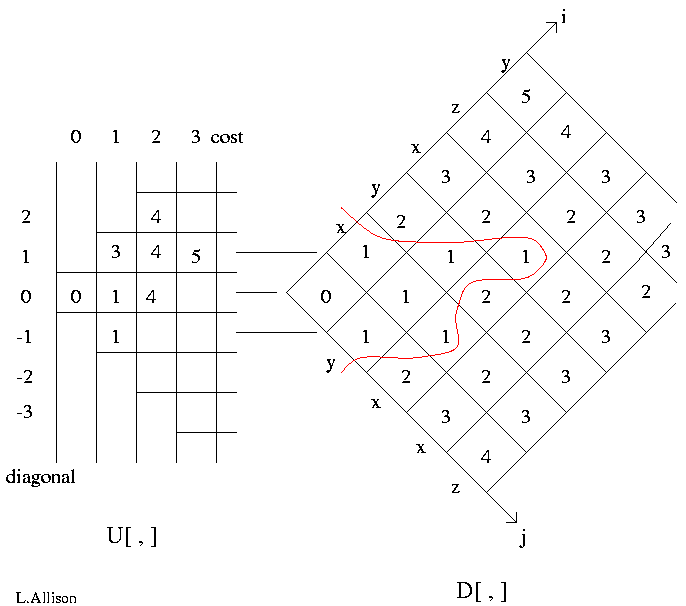
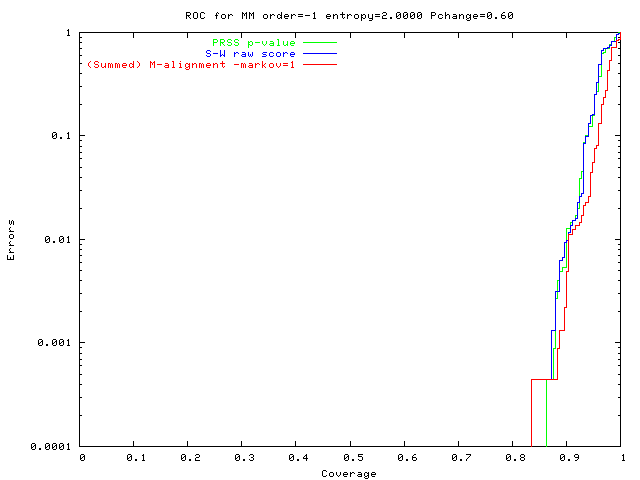
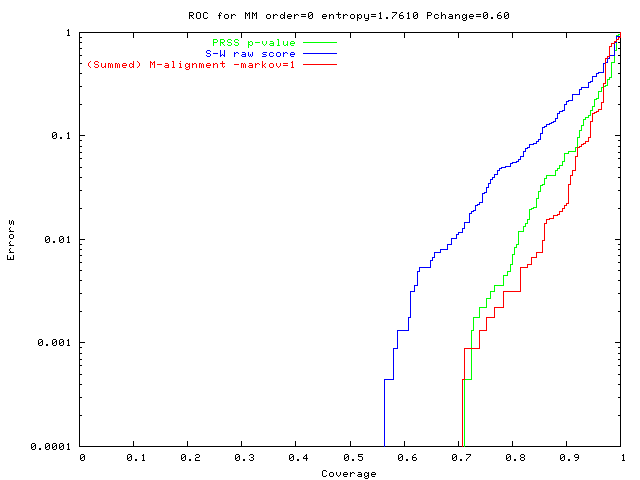
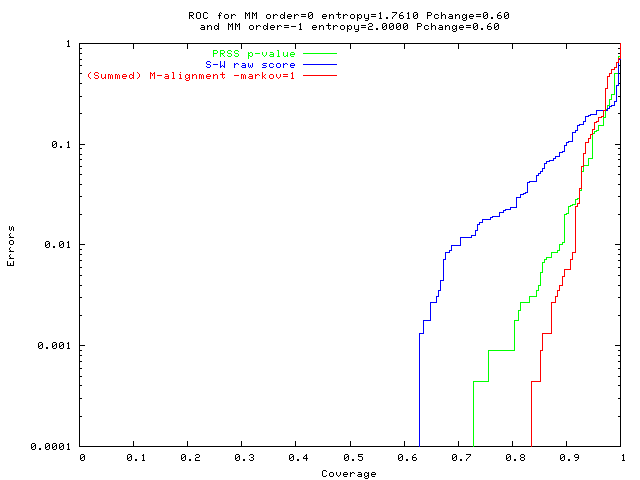
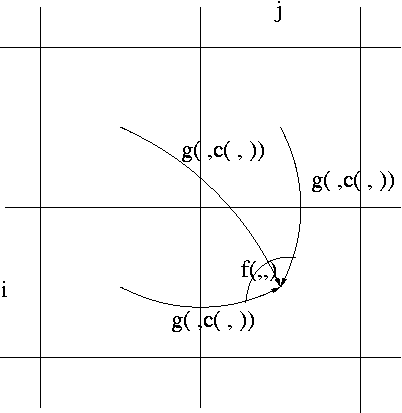An alignment is a hypothesis about how 2 seqs.
are related, if they are related.
It answers the question,
"how did the seqs. evolve, if they are related?"
This is more detailed than the Q., "are the seqs. related?"
The sum-of-probabilities of all alignments answers the latter.
NB. In the "grey zone",
2 distant seqs. may be related even if no one alignment is
an acceptable hypothesis.
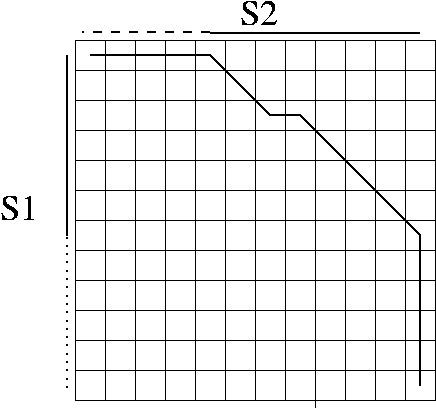
. . . information and local alignment
s1 = α β γ
s2 = δ β' ε,
α γ δ ε about 2bits/base (DNA),
β ~ β',
β+β' about 1bit/base if v.similar.
Must state lengths |α|, |β|, etc..
. . . information and sub-sequence matching
s2 = α s1' β,
α β about 2bits/base (DNA),
s1 ~ s1',
s1+s1' about 1bit/base if v.similar.
. . . information and overlaps
s1 = α β
s2 = γ α',
β γ about 2bits/base (DNA),
α ~ α',
α+α' about 1bit/base if v.similar.
. . . information and parameters
A 5-state mutation model [AWY92] is more complex
than a 3-state model which is more complex than
a 1-state model [BT86].
All parameter values are part of any hypothesis that
two, or more, sequences are related.
They must be paid for to prevent overfitting.
[MML] shows how to do this.
. . . information and lengths
Must state all lengths that are part of any hypothesis,
unless they are common knowledge --
many plausible prob. distributions for such
[integer≥0]
values.
Null hypothesis (seqs. unrelated):
State |s1| and |s2|, or
equivalently |s1|+|s2| and |s1|-|s2|,
the last is expected to be
small +ve or -ve, mean 0,(e.g., can use a code based
on the binomial).
Global alignment (hence seqs. related):
State |alignment| (typically > |s1|, |s2|).
Local alignment, sub-sequence, and overlap problems
have more lengths to state.
Alphabet
Small alphabets, DNA, RNA,
many "chance" symbol matches, e.g., [Dek83] found
lower & upper bounds of 0.5454 & 0.7181 on the (fractional)
|LCS(s1,s2)| for 2 unrelatedseqs. and
an alphabet of size 4.
Can reduce this effect by "k-tupling" symbols of a sequence,
e.g., [WL83],
also see
[HS77] and [Epp92].
Amino acids (20)
skew & bio-chem. properties,
a 20×20 substitution matrix is necessary.
Because of "similar" amino acids,
k × = may be too strong a condition.
Unbounded alphabets, ints, reals
3-D structure . . .
. . . alphabet, 3-D structure
An orientation-independent representation of structure helps:
Protein tertiary structure as set of
points [NW91],
O(|s|4)-time, although can reduce to O(|s|3)
with some loss of info.,
adapts geometric hashing of sets of points.
Protein tertiary structure as
sequence of points [TO89],
DPA, need a c(s1[i], s2[j]) =
(i) ∑m=-n,+n f(|s1[i]-s1[i+m]|, |s2[j]-s2[j+m]|),
takes O(|s1|.|s2|.n)-time,
(ii) DPAi',j'(f(|s1[i]-s1[i']|, |s2[j]-s2[j']|))
--!
the straightforward implementation
takes O(|s1|2.|s2|2)-time.
Mutation costs
Integers,
particularly small integers as in LCS, edit-distance,
In fact can do integer × in
O(n . log(n) . log(log(n)))-time [SS71].
Similar ideas, but replacing (×,+) with (=,+),
solve substring problem in
O(log|alphabet|.n.log(n)2.(log(log(n)))-time [FP74].
Multiple Alignment of k sequences, k≥3.
All pairs:
Alignment {cost | score} is summed
over all 2-sequence projections.
Star based:
Alignment {cost | score} is summed
from the central hypothetical "ancestor" to
each given sequence.
Tree based:
Alignment {cost | score} is summed
over edges of {given | inferred} tree.
Given sequences are leaves of tree.
Internal nodes ~ hypothetical ancestors.
(Probabilities are summed over all possible values at internal nodes.)
. . .
O( |s|k . states2 )-time in general.
NB. k=3 is a useful special case --
each internal node in a phylogenetic-tree has 3 neighbours.
. . . multiple, complexity of,
|s|k = 1,000,000,000, say,
k
2
3
4
6
|s|
32,000
1,000
180
32
The brute-force algorithm is impractical unless
k is small, or
|s| is small,or both.
2-way alignments may somewhat limit the k-dimensional space
to be searched[AL89]
but very quickly heuristics and/or stochastic methods are required.
. . . multiple,
tree-based multiple alignment is based on evolution,
Introduced idea of modelling alignment
(here of a 1-state edit machine);
also see [PAD04] which
generalized the idea.
[AW94] L. Allison, C. S. Wallace,
The posterior probability distribution of alignments and its application
to parameter estimation of evolutionary trees and to optimisation of
multiple alignments.
Samples alignments from their
posterior probability distribution.
Firstly applied to the estimation of the edges of a given
evolutionary tree over several sequences.
Secondly, used in conjunction with simulated annealing,
it gives a stochastic search method for
an optimal multiple alignment.
[AWY92]
L. Allison, C. S. Wallace, C. N. Yee,
Finite-state models in the alignment of macro-molecules.
Sum probabilities of all alignments for
1-, 3-, and 5-state FSAs (HMMs); alignment density plots;
parameter costs by MML prevent overfitting.
[AL89] S. F. Altschul, D. J. Lipman,
Trees, stars and multiple biological sequence alignment.
SIAM J. Appl. Math., 49(1), pp.197-209, Feb. 1989.
Uses pairwise alignments to place an upper bound on the
projections of the optimal multi-alignment onto each pair thus restricting
the volume in the k-dimensional lattice that need be searched for that
optimal k-way alignment.
more . . .
. . . references
[Bel57] R. E. Bellman, Dynamic Programming.
Princeton University Press, 1957.
D.P. in general, not for bioinformatics, e.g.,
the DP paradigm has been used for shortest-paths and minimum spanning tree
problems in graphs, segmentation of time-series, polygon fitting,
and optimal text layout.
[BT86] M. J. Bishop, E. A. Thompson,
Maximum likelihood alignment of DNA sequences.
J. Mol. Biol., 190, pp.159-165, 1986.
Probability based, 1-state.
[Dek83] J. Deken,
Probabilistic behaviour of longest-common-subsequence length.
In Time Warps, String Edits and Macromolecules,
Addison-Wesley, pp.359-362, 1983.
Analysed effect of alphabet size on
chance symbol matches between two sequences.
[Epp92] D. Eppstein, Z. Galil, R. Giancarlo, G. F. Italiano.
Sparse dynamic programming I: linear cost functions.
Includes alignment in
O(|s1|+|s2|+r.log(log min(r,|s1|.|s2|/r)))-time,
where r≤|s1|.|s2| is # of matching fragments found
(and log always +ve). Fast if r is small.
Also see [HS77].
Also part II: convex and concave cost functions, pp.546-567.
[FP74] M. J. Fischer, M. S. Paterson,
String matching and other products.
Sub-string matching with a
don't care (wildcard) symbol,
O(log(|alphabet|).n.log(n)2.log(log(n)))-time,
i.e. nearly linear.
Related to fast integer multiplication.
[Got82] O. Gotoh,
An improved algorithm for matching biological sequences.
J. Mol. Biol., 162, pp.705-708, 1982.
Linear "gap costs".
[Hir75] D. S. Hirschberg,
A linear space algorithm for computing maximal common subsequences.
[MP83] W. J. Masek, M. S. Paterson,
How to compute string-edit distances quickly.
In Time Warps, String Edits and Macromolecules,
Addison Wesley, pp.337-349, 1983.
For a finite alphabet, O(n*n/log(n))-time alg.,
beats O(n2) if n>200,000.
Of theoretical interest only.
[NW91] R. Nussinow, H. J. Wolfson,
Efficient detection of three-dimensional structural motifs in biological
macromolecules by computer vision techniques.
Proc. Natl. Acad. Sci. USA, 88, pp.10495-10499, Dec. 1991.
Related to geometric hashing:
Any set of 3 residues defines a coordinate system (CS).
For each candidate seq., for each such CS, hash (bucket) every residue.
Count the hash "votes."
[PAD99]
D. R. Powell, L. Allison, T. I. Dix,
A versatile divide and conquer technique for optimal
string alignment.
Reduces space complexity to
linear in string length for 2 sequences, quadratic for 3 sequences, etc.,
easy to use with complex indel/gap-costs.
Can also be used with fast [Ukk82]-style algorithms.
[PAD00]
D. R. Powell, L. Allison, T. I. Dix,
Fast, optimal alignment of three sequences using linear gap costs.
Aligns non-random sequences,
builds seq. model into DPA, global and local-alignment,
optimal and sum of probabilities, inc. linear gap costs.
(Also see [APD99].)
[TO89] W. R. Taylor, C. A. Orengo,
Protein structure alignment.
Compares protein structures based on
distance plots.
[Ukk83] E. Ukkonen,
On approximate string matching.
Proc. Int. Conf. on Foundations of Computation Theory,
LNCS 158, pp.487-495, Aug. 1983,
doi:10.1007/3-540-12689-9_129.
Edit dist., fast,
O(d×n)-time worst case, O(n+d2) on average.
[WL83] W. J. Wilbur, D. J. Lipman,
Rapid similarity searches of nucleic acid and protein banks.
Proc. Natl, Acad. Sci. USA, 80, pp.726-730, Feb. 1983,
@pnas
Makes k-tuples whuch increases the effective alphabet size
by a power of k. This gives a small number of cross-sequence matches,
so a faster algorithm (~Hirschberg?) can be used.
Tupling, aka "word", "k-mers" etc., later used in BLAST and
other such programs.
[YA93] C. N. Yee, L. Allison,
Reconstruction of strings past.
Use of single optimal alignment gives biased estimates of
the evolutionary "distance" between two strings but the r-theory,
average over all alignments, recovers accurate estimates over
a very wide range of similarity.